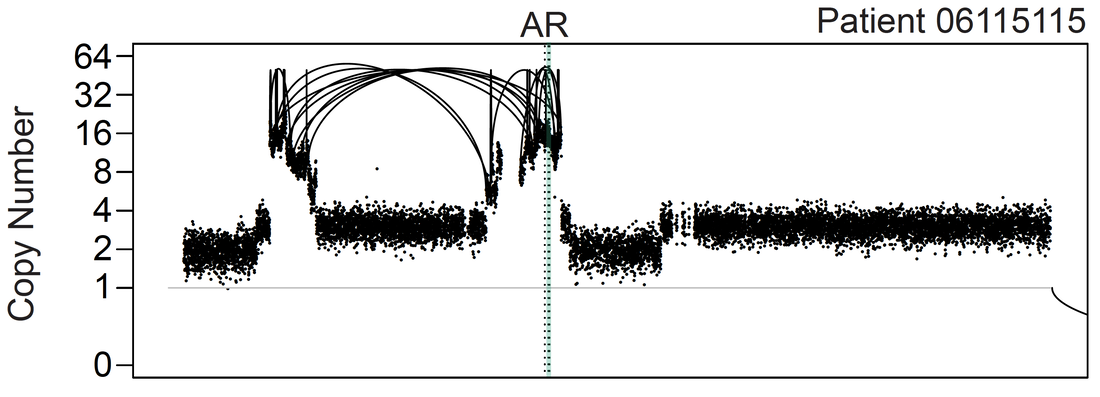
Complex rearrangement of the AR locus visualized by linked-read sequencing.
Viswanathan, et al., Cell, 2018. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing
Viswanathan, et al., Cell, 2018. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing
Cancer Genomic Technology
As part of the continuing revolution in genome technology, the Meyerson laboratory has developed powerful experimental and computational methods to bring new technologies to cancer research. We were the first group to use single nucleotide polymorphism (SNP) arrays for analysis of cancer loss of heterozygosity (Lindblad-Toh et al., 2000) and for copy number analysis, leading to our selection as the leaders of the Genome Characterization Center for copy number for The Cancer Genome Atlas (TCGA). Later, we performed the first next-generation sequencing analysis of cancer mutations (Thomas et al., 2006), demonstrating how digital sequencing could reveal cancer heterogeneity.
Among our innovations in computational analysis, members of the Meyerson group, with our collaborators, have developed computational methods to determine absolute cancer copy number in the face of heterogeneity (Carter et al., 2012), identify active retro-transposition in cancer (Helman, et al., 2014), and determine tumor fraction in cell-free DNA from ultra low-pass genome sequencing (Adalsteinsson, et al, 2017).
Our current technology efforts are focused on long-read and linked-read sequencing methods to identify rearrangements across repetitive elements. Using these methods we have recently identified duplications of lineage-specific super-enhancers near the androgen receptor oncogene in prostate carcinoma (Viswanathan, et al, 2018).
As part of the continuing revolution in genome technology, the Meyerson laboratory has developed powerful experimental and computational methods to bring new technologies to cancer research. We were the first group to use single nucleotide polymorphism (SNP) arrays for analysis of cancer loss of heterozygosity (Lindblad-Toh et al., 2000) and for copy number analysis, leading to our selection as the leaders of the Genome Characterization Center for copy number for The Cancer Genome Atlas (TCGA). Later, we performed the first next-generation sequencing analysis of cancer mutations (Thomas et al., 2006), demonstrating how digital sequencing could reveal cancer heterogeneity.
Among our innovations in computational analysis, members of the Meyerson group, with our collaborators, have developed computational methods to determine absolute cancer copy number in the face of heterogeneity (Carter et al., 2012), identify active retro-transposition in cancer (Helman, et al., 2014), and determine tumor fraction in cell-free DNA from ultra low-pass genome sequencing (Adalsteinsson, et al, 2017).
Our current technology efforts are focused on long-read and linked-read sequencing methods to identify rearrangements across repetitive elements. Using these methods we have recently identified duplications of lineage-specific super-enhancers near the androgen receptor oncogene in prostate carcinoma (Viswanathan, et al, 2018).